Generalidades
La Distrofia
Muscular de Duchenne (DMD) –pseudohipertrófica- es la más común y la más rápidamente
progresiva de este grupo de trastornos neuromusculares.
La
enfermedad se hereda como un rasgo recesivo ligado al sexo; el 50% de los hombres
son afectados y el 50 % de las mujeres son portadoras. La biopsia muscular es
anormal en la tercera parte de los portadores.
Aproximadamente
el 75% de los portadores pueden ser descubiertos por una elevación de las
enzimas séricas, especialmente la CPK.
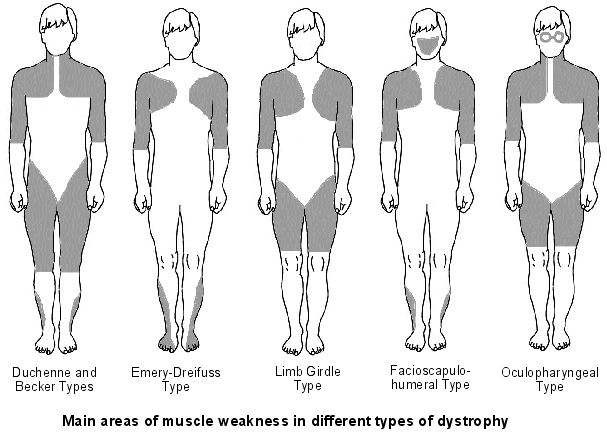
Las
Distrofias musculares de Duchenne (DMD) y Becker (DMB) tienen signos y síntomas
similares y son causadas por diferentes mutaciones en el mismo gen. Porque estas distrofias musculares resultan de una
disrupción del complejo distrofina-glicoproteínas (DAP), estas condiciones son
clasificadas como Distrofinopatías.
Hallazgos clínicos
Tiene
una frecuencia elevada, de 1:3.500. Comienza entre los 2 y los 4 años con
retraso motor (40%), marcha anormal (30%),
trastorno del lenguaje y el habla (8%).
El
desarrollo motor inicial es poco notorio, y el paciente llega a caminar varios meses
mas tarde con relación a sus hermanos. Los padres generalmente comienzan a
preocuparse cuando notan que el niño presenta torpeza y se cae con frecuencia.
El niño no aprende a correr o a subir escaleras. Hacia los 3 años de edad se le
aprecia aumento del volumen de las pantorrillas: “Pseudohipertrofia”.
Pseudohipertrofia
gemelar
El
niño puede presentar dificultad para entrar a un automóvil y para salir de el.
Hay aumento de la lordosis lumbar y “marcha anadina”
El
acortamiento aquiliano bilateral ocasiona cierto grado de equinovaro. Los músculos
proximales alrededor de las cinturas escapular y pélvica son afectados por una notoria
debilidad. El niño es incapaz de participar en ninguna clase de actividad atlética.
La discapacidad empeora progresivamente y hacia los 10 años de edad el niño
puede estar limitado a una silla de ruedas.
La
atrofia muscular frecuentemente acompaña a la grave debilidad de los músculos
proximales. El paciente trata de levantarse del piso en forma característica, “trepando
por los muslos con los brazos” (Signo de
Gower), debido a la debilidad de los extensores y abductores de cadera.
Al
comienzo los reflejos profundos están normales; sin embargo, a medida que el
trastorno aumenta, disminuyen o desaparecen, con excepción de los reflejos
aquilianos. En seguida se presenta un acortamiento de los tendones de Aquiles y
contracturas en las rodillas y los codos. Para la época de la adolescencia, la
mayor parte de los pacientes están limitados al lecho. En último termino,
fallece por complicaciones bronconeumónicas o infecciones del tracto urinario.
Aproximadamente
el 30% de los niños con DMD pueden tener retardo mental, lo cual es indicativo
de un compromiso sistémico.
Exámenes complementarios
Los estudios enzimáticos demuestran un
marcado aumento de la creatinfosfoquinasa (CPK) en las etapas iniciales de la
enfermedad, el cual se hace menos notorio a medida que el cuadro clínico va
aumentando. Otras anormalidades enzimáticas comprenden la elevación de la
aldolasa, de la deshidrogenasa láctica (DHL) y de la transaminasa glutámica
oxalacética (TGO) séricas.
Los estudios electromiográficos están caracterizados
por un trazado miopático, que consiste en potenciales polifásicos de bajo
voltaje, de duración anormalmente breve.
Anormalidades electrocardiográficas se
pueden encontrar desde el comienzo de la enfermedad. Las cardiomiopatías y la
insuficiencia cardíaca congestiva (ICC)
son complicaciones frecuentes que se presentan durante la tercera década de la
vida.
En la biopsia muscular se puede apreciar un
deterioro de las fibras musculares, migración de los núcleos del sarcolema al
centro de las fibras musculares, reemplazo por grasa y fragmentación y proliferación
del tejido conjuntivo.
Tratamiento
El
tratamiento farmacológico (corticoterapia: prednisona y deflazacort) se
utilizan actualmente en la DMD y han demostrado alterar la historia natural de la
DMD (Liew W, Kang P. 2013)). Un programa de fisioterapia de ejercicios activos
regulares pero moderados, y de diversas técnicas de elongación, pueden prevenir
las contracturas.
La
regla de oro con relación a la cirugía en las distrofias musculares es su objetivo funcional. Puede
prolongarse la ambulación con la liberación quirúrgica de la bandas
iliotibiales y del manejo del acortamiento del tendón aquiliano.
Es recomendable
que el paciente participe en actividades con otros niños y se debe estimular la
educación dentro de un programa regular, siempre que ello se posible.
Nuestro
esfuerzo para clarificar el diagnóstico específico de distrofia muscular en
cada paciente tiene por objeto definir el plan terapéutico, establecer el
pronóstico, y brindar consejo genético
adecuado a la familia.
Referencias:
1.
Jabbour/Duenas. “Manual de Neurología Infantil”. Fondo Educativo Interamericano S.A. 1978
2. Erazo-Torricelli R. “Actualización
en distrofias musculares”. [Rev Neuro. 2004; 39: 860-71]
Adaptado por: Dr. Carlos Arce – Médico Fisiatra –
Hospital Almenara (2014)